Sudden Arrhythmias – Causes of SADS – Sudden arrhythmic death syndrome
The conditions responsible for SADS cause a cardiac arrest by bringing on a ‘ventricular arrhythmia’ (a disturbance in the heart’s rhythm), even though the person has no structural heart disease.
There is a group of relatively rare diseases called ion channelopathies that affect the electrical functioning of the heart without affecting the heart’s structure. This means that they can only be detected in life and not at post-mortem. Ion channelopathies are probably responsible for 4 in every 10 cases of Sudden Arrhythmia Death Syndrome. There are several different types of ion channelopathies including:
-
Long QT Syndrome (LQTS)
-
Brugada Syndrome
-
CPVT (catecholaminergic polymorphic ventricular tachycardia
-
PCCD (progressive cardiac conduction defect)
-
IVF (idiopathic ventricular fibrillation), and
-
sodium channel disease.
Structural heart disease is occasionally found to be a cause of SADS (between 1 and 2 in every 10 cases).
Ion channelopathies
Ion channelopathies are rare genetic conditions that are caused by abnormalities of the ‘DNA’ known as ‘mutations’. They are usually inherited from parents although they can occur for the first time in a family. If they occur for the first time they are described as ‘sporadic’.
The mutations affect certain genes – specific segments of the DNA that are responsible for the production of cardiac ‘ion channels’. An ‘ion’ is a chemical substance – such as sodium or potassium – that carries an electrical charge and forms the basis of the movement of electricity through the heart muscle. An ‘ion channel’ is the route that the ions take in and out of the heart muscle cells to allow the movement of electricity. The ion channels regulate the flow of electrical charge.
If these channels do not behave normally, the electrical function of the heart becomes abnormal. The person can then be prone to arrhythmias (disturbances in the heart’s rhythm) that can cause blackouts, cardiac arrest and in some cases sudden death.
Below we describe the different types of channelopathies, the tests needed to diagnose them and the treatment that may be needed for each one.
Long QT Syndrome (LQTS)
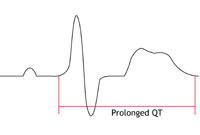 LQTS is the most common and best understood type of channelopathy. It occurs in about 1 in 5,000 people. In 7 in every 10 people with LQTS, the ion channels involved have been identified. In most cases two of the potassium channels that regulate the movement of potassium ions from the inside to the outside of the cell are affected. In a small proportion of people with LQTS, a sodium channel that regulates t
LQTS is the most common and best understood type of channelopathy. It occurs in about 1 in 5,000 people. In 7 in every 10 people with LQTS, the ion channels involved have been identified. In most cases two of the potassium channels that regulate the movement of potassium ions from the inside to the outside of the cell are affected. In a small proportion of people with LQTS, a sodium channel that regulates t
he flow of sodium ions from the outside to the inside of cells is affected.
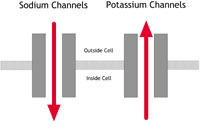
In people with potassium channel associated LQTS, the channels do not behave as efficiently as normal. They let potassium ions into the cell too slowly. If the sodium channel is affected, too many sodium ions are allowed into the cell. (See the LQTS diagram – figure 2B – below.) This results in an electrical disturbance in the cells of the
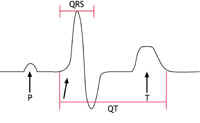
heart called ‘prolonged repolarisation’. This can be seen on an ECG recording as a lengthening of the time period known as the ‘QT interval’. This is where the name Long QT Syndrome comes from.
 Rare forms of LQTS known as Andersen’s and Timothy Syndromes have been associated with potassium and calcium channel abnormalities respectively.
Rare forms of LQTS known as Andersen’s and Timothy Syndromes have been associated with potassium and calcium channel abnormalities respectively.
What are the symptoms?
LQTS varies greatly in severity. Symptoms vary according to the type of channel involved, whether the person is male or female, their age, and the length of the QT interval on the ECG. Males are more likely to have symptoms before puberty, while females are more likely to have them in adolescence and early adulthood. Relatives from the same family who have inherited the same mutation may have very different experiences. For example, some may have a normal QT interval and not have any symptoms; some may have a very abnormal QT interval but no symptoms; and some may have a very abnormal QT interval and have many events that put them at risk.
The most common symptom of LQTS is blackouts. Sometimes palpitations due to extra or ‘ectopic’ heartbeats can be a problem.
Potassium channel LQTS is associated with sudden death which is related to exercise or when the person has been startled or awoken suddenly (‘sudden arousal’). The sodium channel form is associated with death while asleep.
Are there any physical signs?
There are no physical signs of LQTS. However, people with Andersen’s Syndrome may also have muscle weakness or minor abnormalities of the skull, chin, fingers and toes.
How is it diagnosed?
Diagnosis involves having an ECG. Sometimes it is possible to tell which ion channel has been affected just by looking at the ECG recording. Unfortunately, in many people who might be carriers, the ECG does not show any sign of the condition. Repeated ECGs, exercise tests and 24-48 hour tape monitoring may be needed before any hint of the condition is seen, and even then there may be no sign of it (we describe all these tests in cardiac tests).
Genetic testing can sometimes identify carriers of LQTS. Unfortunately, this form of testing is limited at the moment, as 3 in every 10 people who are known to have LQTS do not have mutations of the genes known to be associated with LQTS. An additional problem is that most families who do have the mutations appear to have a specific change to the DNA code which is not found in other families (known as a ‘private’ mutation). This sometimes makes it difficult to decide whether a mutation is causing the disease or not. Things are further complicated by the fact that people with the same mutation can have effects that vary greatly in severity. All of this makes it very difficult for doctors to decide on the best way to treat people with this condition.
Treatment and advice
If you have LQTS, your doctor will advise you to avoid excessive exercise or strenuous athletic activities. He or she will also advise you to avoid certain drugs that can make the condition worse and which could increase the risk of blackouts and sudden cardiac death (see lifestyle advice).
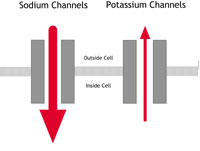
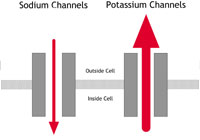
| A: Normal heart | B: LQTS | C: Brugada Syndrome or PCCD |
 |
|
 |
|
Figure 2A-C: What happens in ion channelopathies |
||
|
A In a normal heart |
B In people with LQTS |
C In people with Brugada Syndrome or PCCD |
The level of risk of sudden death helps decide on the need for treatment. Those who are statistically at greatest risk of sudden death are people with one or more of the following features:
-
a previous cardiac arrest
-
blackouts
-
a very long QT interval on the ECG
-
sodium channel mutations
-
young adult women.
Children who are most at risk tend to be young boys before puberty, and girls who are passing into puberty.
Drugs
The first line of treatment is with drugs. The most commonly used drugs are betablockers. These block the effects of adrenaline and associated natural chemicals in the body that make the heart pump harder and faster. They therefore also block the effects of exercise on the heart. They are effective in the most common forms of LQTS as they reduce symptoms and the risk of sudden death. However, they are less effective in people with the sodium channel form of LQTS.
There are other more recent trends in drug treatment that look promising, but their long-term benefits are unknown. These involve using antiarrhythmic drugs. These drugs block disturbances in the heart rhythm that can cause sudden death. Potassium supplement pills have also been tried with occasional success.
Pacemaker or ICD
If you are at high risk (for example if you have already had a cardiac arrest), or if drugs have failed to control your symptoms, your doctor may advise you to have a pacemaker or an implantable cardiac defibrillator (ICD) fitted, as well as taking your medication. A pacemaker and an ICD both consist of an electronic box that is inserted under the skin and attached to the heart by special electrical ‘leads’. A pacemaker controls the heart rate and stops any excessive slowing of the heart that could trigger an arrhythmia. An ICD acts in the same way as a pacemaker but it can also identify any dangerous arrhythmias and deliver an electrical shock to reset the heart (for more information on pacemakers and ICDs see technical terms).
Surgery
Another option is to perform surgery to disrupt the nerves that release adrenaline and related chemicals at the heart. This is known as ‘cervical sympathectomy’ and involves operating on the left side of the neck.
Brugada Syndrome
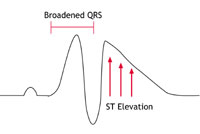
This condition was first identified in the early 1990s. It is an uncommon condition in the western world but seems to be much more common among young men in South East Asia. In the western world it affects mainly young and middle-aged adult men. It has been associated with mutations in the same sodium channel that is affected in LQTS, but this appears to account for only 1 in every 5 people with the condition. The sodium channel behaves abnormally in that movement of sodium ions into the cells is restricted. This results in particular changes on the ECG (as shown below in figure 3C) but no abnormalities in the structure of the heart.
|
A: Normal |
B: LQTS | C: Brugada Syndrome. |
 |
|
 |
|
Figures 3A-C: A simplified version of the ECG as it would appear in one lead in the following circumstances: |
||
What are the symptoms?
Some people with Brugada Syndrome may have no symptoms at all. In others, the most common symptoms are blackouts. Some people may notice palpitations due to ectopic beats. Sudden death may occur. If it does, it usually happens while the person is sleeping.
Are there any physical signs?
There are no associated physical signs.
How is it diagnosed?
Diagnosis involves having an ECG. The changes characteristic of Brugada Syndrome may appear on the ECG continuously or intermittently, or they may not show at all. Sometimes the presence of a fever can bring out the ECG changes. If they do not show up on the ECG, there are tests that can make the ECG changes visible. These are called ‘provocation tests’ and involve having a short injection of an antiarrhythmia drug while you are having an ECG (see cardiac tests). The drugs most commonly used for this are ajmaline and flecainide. There is some controversy, however, about how much reassurance a negative result should give. Researchers have found that, in some carriers who have already been identified by genetic testing,changes on the ECG are not seen even with a provocation test. However, in these people the level of risk does appear to be low.
Genetic testing is not very useful for diagnosing Brugada Syndrome because mutations have been found in only a small proportion of people known to have the syndrome.
Treatment and advice
The outlook for people with Brugada Syndrome can be poor, especially in people who get symptoms or have already had a cardiac arrest, the highest rates of sudden cardiac death being found among young male adults. It is therefore standard practice for high risk carriers to have an ICD fitted as this is very successful form of protection. For more information on ICDs see technical terms. To date medication has not been shown to be protective in people with Brugada Syndrome.
Unfortunately it can be very difficult for doctors to decide how to treat those people who do not get symptoms but who have an abnormal ECG. An EPS (an electrophysiological study) may help to identify those people who do or do not need an ICD. Research has suggested, however, that people with normal ECGs and no symptoms should be safe without any treatment. It is unusual for children to be at high risk. Carriers are advised to take fever lowering drugs such as paracetamol or ibuprofen due to the potential effect of fever on the ECG.
CPVT (Catecholaminergic polymorphic ventricular tachycardia)
CPVT is a rare condition found in young people and children. It causes a particular type of arrhythmia. It has been associated with two genes that make proteins found inside the cell – the human ryanodine receptor (a calcium ion channel) and calsequestrin (a protein that interacts with the channel). These regulate the release of calcium ions into the rest of the cell. If these do not function normally, the level of calcium inside the cell becomes too high, resulting in the arrhythmias characteristic of CPVT.
What are the symptoms?
Some people with CPVT have no symptoms at all. Others may have blackouts. Sudden death may occur while the person is exerting themselves or suffering emotional stress.
The condition can affect children and seems to cause more blackouts in males than in females.
Are there any physical signs?
There are no physical signs.
How is it diagnosed?
The diagnosis is usually made after the chance recording of arrhythmias that are characteristic of CPVT, while the person is doing exercise. Genetic testing is also useful in cases where a member of the same family has already been found to carry a mutation and is showing the signs of the condition.
Treatment and advice
Your doctor will advise you to take beta-blockers (a type of drug) and to restrict the amount of exercise you do. This combination greatly improves the outlook for people with CPVT. About 1 in every 3 people with the condition will also need to have an ICD fitted (for more on ICDs see technical terms).
PCCD (Progressive cardiac conduction defect)
PCCD is a rare condition. In people with PCCD, the heart’s electrical impulses are conducted very slowly and this results in the gradual development over time of ‘heart block’. (Heart block is a failure of the heart’s electrical impulse to conduct properly from the top chambers [the atria] to the bottom chambers [the ventricles]. The severity of the condition and its associated risk can vary.) PCCD can cause arrhythmias – either because the heart’s rhythm is too sluggish (bradycardia and asystole), or because of rapid rhythm disturbances (tachycardia) arising from parts of the heart that have escaped normal regulation. In some people PCCD has been associated with sodium channel mutations that cause changes in channel behaviour similar to those found in people with Brugada Syndrome (see the Brugada Syndrome diagram – figure 2C – above).
What are the symptoms?
Dizziness and blackouts are the usual symptoms. Sudden death may also occur.
Are there any physical signs?
There are no physical signs.
How is the diagnosis made?
The ECG abnormalities may be detected either on a standard ECG or with Holter monitoring. An electrophysiological study may also help the doctor make a diagnosis. (We describe all these tests in cardiac tests.) If a sodium channel mutation is identified in affected members of a family then it may also be found in other relatives.
Treatment and advice
If you have PCCD you will need to have a pacemaker fitted in order to stop dangerous bradycardia from occurring. This may not prevent ‘escape tachycardias’ so you may also need to take antiarrhythmic drugs. Some people may need to have an ICD fitted instead of a pacemaker (for more on pacemakers and ICDs go to technical terms). Medication alone does not help.
IVF (Idiopathic ventricular fibrillation)
This term describes the group of conditions responsible for life-threatening, rapid rhythm disturbances without any signs of heart disease. Brugada Syndrome and CPVT form part of this group but there have been reports of patients with IVF who do not have the ECG changes characteristic of the Brugada Syndrome but who do have sodium channel mutations. Treatment includes having an ICD fitted, and can be successful in protecting the person.
Sodium channel disease
There are very rare and specific sodium channel mutations that can cause Long QT Syndrome, Brugada Syndrome and/or PCCD in the same family. They can be diagnosed and treated as described above and can be identified by genetic testing.
Structural heart disease
In some cases, the pathologist cannot confirm a diagnosis of structural heart disease – either because there is no evidence of it, or because there is not enough evidence and the heart is felt to be relatively normal. So the death will be recorded as SADS. This may happen even in cases where evidence of inherited structural heart disease is subsequently detected in other members of the victim’s family. The presence of very subtle structural heart disease in the victim may, however, have been enough to cause sudden cardiac death.
In these circumstances the most common causes of death are:
-
arrhythmogenic right ventricular cardiomyopathy (ARVC)
-
dilated cardiomyopathy (DCM)
-
hypertrophic cardiomyopathy (HCM)
-
mitral valve prolapse (MVP)
-
Wolff-Parkinson-White Syndrome (WPW).
We explain each of these in sudden death.

